サンプル内の転写物を定量するときに、RT-PCR (real time PCR) などが一般的に利用されている。RT-PCR では、サンプル内に定量対象の cDNA とコントロールとなる cDNA (16S rRNA など)を加えて、PCR により増幅する。PCR が 1 サイクルごとに cDNA の量が 2 倍に増える。これを繰り返すことで、cDNA の量が指数関数的に増幅される。やがて、あるサイクルで定量対象の転写物か、あるいはコントロールの転写物が、機械の検出限界を超えて、初めて機械に検出される。この値が CT である。サンプル内に含まれている cDNA の量が多ければ、より少ない PCR サイクルで検出される。逆に、サンプル内に含まれている cDNA の量が少なければ、より多くの PCR サイクルを必要とする。定量対象転写物の CT とコントロール転写物の CT を比べることで、定量対象転写物の相対発現量や、fold-change などを計算できる。
ここで、CT 値から fold-change を求める方法や R でグラフを描く方法などを紹介する。例として取り上げたデータや例などは RT-PCR の実験プロトコルについて述べた論文に使われている値を利用した (Schmittgen et al., Nat Protoc, 2008)。
処理後の fold-change の計算方法
RT-PCR は、一般に、定量対象の 1 遺伝子(転写物)について 1 cDNA ライブラリーを準備し、次にテクニカルエラーを防ぐために、準備した cDNA ライブラリーを 3 つに分けて RT-PCR を行う。つまり、techinical replicate が 3 つ存在し、それぞれの technical replicate から CT 値が得られる。この場合、fold-change を計算するときは、これらの値を平均してから扱う。
処理後発現量が増加した場合の fold-change の計算例
HOXD10 遺伝子について、処理前と処理後の発現量を RT-PCR で定量して、fold-chang を求める例を示す。HOXD10 遺伝子およびコントロール 18S rRNA の CT 値を表中に示した値とする。
| 処理前 | 処理後 | |||||
| technical replicate | 1 | 2 | 3 | 1 | 2 | 3 |
| HODX10 | 27.5 | 27.4 | 27.6 | 24.6 | 24.4 | 24.8 |
| 18S rRNA | 9.7 | 9.8 | 9.9 | 9.9 | 9.9 | 9.9 |
処理後の HODX10 の発現量は、処理前に比べどのぐらい変化したかを表す fold-change を求める。
- 各 technical replicate から計算された CT の平均を計算する。
- 処理前と処理後それぞれについて、HODX10 の CT の平均から 18S rRNA の CT を引く。(ΔCT)
- 処理前の ΔCT から処理後の ΔCT を引く。(ΔΔCT)
- - ΔΔCT をべき指数として、2 のべき乗を求める。この値が、処理前に対する処理後の fold-change になる。
R を利用して以下のように計算できる。この例では、処理後は処理前に比べ 8 fold-changes となる。
hodx <- data.frame(untreat = c(27.5, 27.4, 27.6),
treat = c(24.6, 24.4, 24.8))
ctrl <- data.frame(untreat = c(9.7, 9.8, 9.9),
treat = c(9.9, 9.9, 9.9))
# 1. calculate average
hodx.ave <- colMeans(hodx)
ctrl.ave <- colMeans(ctrl)
# 2. calculate delta Ct
deltaCt <- hodx.ave - ctrl.ave
# 3. calculate delta delta Ct
deltadeltaCt <- deltaCt[2] - deltaCt[1]
# 4. caculate foldchange
2 ^ (-deltadeltaCt)
## [1] 8
処理後発現量が増加した場合の計算方法
次に、処理後が処理前に比べて down-regulated されている例を示す。HOXD10 遺伝子およびコントロール 18S rRNA の CT 値を表中に示した値とする。
| 処理前 | 処理後 | |||||
| technical replicate | 1 | 2 | 3 | 1 | 2 | 3 |
| HODX10 | 24.6 | 24.4 | 24.8 | 27.5 | 27.4 | 27.6 |
| 18S rRNA | 9.9 | 9.9 | 9.9 | 9.7 | 9.8 | 9.9 |
R を利用して fold-change を計算する。この場合、fold-change が 1 よりも小さい数値を取っているので、その逆数を計算する。この逆数に -1 をかけた値が fold-change である。この例では処理後は処理前に比べ 8 fold-change も発現量が少ない。
hodx <- data.frame(untreat = c(24.6, 24.4, 24.8),
treat = c(27.5, 27.4, 27.6))
ctrl <- data.frame(untreat = c(9.9, 9.9, 9.9),
treat = c(9.7, 9.8, 9.9))
hodx.ave <- colMeans(hodx)
ctrl.ave <- colMeans(ctrl)
deltaCt <- hodx.ave - ctrl.ave
deltadeltaCt <- deltaCt[2] - deltaCt[1]
2 ^ (-deltadeltaCt)
## [1] 0.125
-1 / (2 ^ (-deltadeltaCt))
## [1] -8
時系列データの fold-change の計算方法
次に、ある処理を加えた後の発現量の変化を時系列的に定量したデータから、fold-change を求める方法およびグラフを描く方法示す。例えば、データは以下のように得られたとする。
| 処理前 (0 hr) | 処理後 (2 hr) | 処理後 (4 hr) | 処理後 (6 hr) | |||||||||
| technical replicate | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 |
| HODX10 | 27.5 | 27.4 | 27.6 | 25.2 | 25.1 | 25.2 | 24.8 | 24.9 | 24.9 | 25.2 | 25.1 | 25.2 |
| 18S rRNA | 9.9 | 9.8 | 9.9 | 9.7 | 9.8 | 9.9 | 9.7 | 9.9 | 9.9 | 9.8 | 9.8 | 9.9 |
まず、上の例と同様に、処理前と処理後の 3 time points とを比べて、fold-change を求める。
hodx <- data.frame(H0 = c(27.5, 27.4, 27.6),
H2 = c(25.2, 25.1, 25.2),
H4 = c(24.8, 24.9, 24.9),
H6 = c(24.6, 24.4, 24.8))
ctrl <- data.frame(H0 = c(9.9, 9.8, 9.9),
H2 = c(9.7, 9.8, 9.9),
H4 = c(9.7, 9.9, 9.9),
H6 = c(9.8, 9.8, 9.9))
hodx.ave <- colMeans(hodx)
ctrl.ave <- colMeans(ctrl)
deltaCt <- hodx.ave - ctrl.ave
deltadeltaCt <- deltaCt - deltaCt[1]
fc <- 2 ^ (-deltadeltaCt)
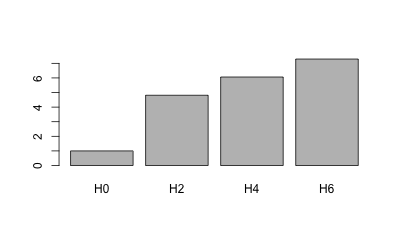
fc
## H0 H2 H4 H6
## 1.0000000 0.7937005 0.6597540 0.1309118
barplot(fc)
biological replicate が存在する場合の fold-change の計算方法
これまでに見た例では 1 つの cDNA サンプルを 3 つに分けて、PCR を行って CT を定量してきた。これが technical replicate に当たる。
次に 3 つのマウスなどから採取したサンプル(biological replicate)を利用した例を示す。まず、biological replicate それぞれ独立しているとみなして、ΔCT を計算し、次に ΔCT から平均と標準偏差を計算する。fold-change (ΔΔCT) は最後に ΔCT の平均から計算する。
| 処理前 | 処理後 | |||||||||||||||||
| biological replicate | 1 | 2 | 3 | 4 | 5 | 6 | ||||||||||||
| technical replicate | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 |
| HODX10 | 27.3 | 27.3 | 27.4 | 27.1 | 27.9 | 27.4 | 27.9 | 27.3 | 27.8 | 24.6 | 24.4 | 24.8 | 24.8 | 24.5 | 24.6 | 24.6 | 24.7 | 24.6 |
| 18S rRNA | 9.9 | 9.8 | 9.9 | 9.7 | 9.8 | 9.9 | 9.7 | 9.9 | 9.9 | 9.7 | 9.7 | 9.9 | 9.7 | 9.7 | 9.7 | 9.9 | 9.8 | 9.9 |
hodx.bf <- data.frame(M1 = c(27.3, 27.3, 27.4),
M2 = c(27.1, 27.9, 27.4),
M3 = c(27.9, 27.3, 27.8))
hodx.af <- data.frame(M4 = c(24.6, 24.4, 24.8),
M5 = c(24.8, 24.5, 24.6),
M6 = c(24.6, 24.7, 24.6))
ctrl.bf <- data.frame(M1 = c(9.9, 9.8, 9.9),
M2 = c(9.7, 9.8, 9.9),
M3 = c(9.7, 9.9, 9.9))
ctrl.af <- data.frame(M4 = c(9.7, 9.7, 9.9),
M5 = c(9.7, 9.7, 9.7),
M6 = c(9.9, 9.8, 9.9))
# before treatment
hodx.ave.bf <- colMeans(hodx.bf)
ctrl.ave.bf <- colMeans(ctrl.bf)
deltaCt.bf <- hodx.ave.bf - ctrl.ave.bf
p2deltaCt.bf <- 2 ^ (- deltaCt.bf)
# after treatment
hodx.ave.af <- colMeans(hodx.af)
ctrl.ave.af <- colMeans(ctrl.af)
deltaCt.af <- hodx.ave.af - ctrl.ave.af
p2deltaCt.af <- 2 ^ (- deltaCt.af)
mean(p2deltaCt.bf)
## [1] 4.869655e-06
sd(p2deltaCt.bf)
## [1] 6.219517e-07
mean(p2deltaCt.af)
## [1] 3.403019e-05
sd(p2deltaCt.af)
## [1] 1.966643e-06
# foldchange
mean(p2deltaCt.af) / mean(p2deltaCt.bf)
## [1] 6.988215
この結果を見ると、処理前の ΔCT 値は 0.00000487 ± 0.00000062 であり、処理後の ΔCT 値は 0.00003403 ± 0.00000197 である。処理前に比べて、処理後の fold-change は 0.00003403 / 0.00000487 = 6.99 と計算される。
References
- Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008, 3(6):1101-8. DOI: 10.1038/nprot.2008.73 PMID: 18546601